With the proposed goals of ‘Carbon peak’ and ‘Carbon neutrality’ of the world, hydrogen energy has become increasingly attention due to its abundant reserves, environmentally friendly characteristics, and high energy density. Utilizing hydrogen as a fuel cell for power generation in automobiles, trains, ships, and aviation is an effective method for reducing carbon emissions. The critical challenge is to find a suitable solid-state hydrogen storage material that has excellent hydrogen storage properties and can release hydrogen under compatible conditions. Referring to the guideline established by the U.S. Department of Energy, an effective solid-state hydrogen storage device should be capable of storing at least 6.5 wt% of hydrogen to achieve the objective of a 500-mile (~805 kilometer) range for a hydrogen fuel cell vehicle. A team led by researcher Qing Peng, the Institute of Mechanics, Chinese Academy of Sciences, and Prof. Yifang Ouyang, Guangxi University has developed a two-dimensional solid-state hydrogen storage material system called Ti-decorated Irida-Graphene (TIG) through first-principles calculations. Irida-Graphene (IG) is composed of three-, six-, and eight-atom carbon rings. They found that by introducing modified titanium (Ti) atoms to IG, its hydrogen storage performance can reach up to 7.7 wt%.
The research team conducted a computational study on the adsorption capacity of titanium atoms at various sites in IG. They found that the most stable adsorption site is the Hollow site located above the six atoms of IG. Binding energy analysis reveals that a single Ti atom in the primitive unit-cell of Ti-decorated Irida-Graphene is capable of binding up with 5 H2 molecules and the average adsorption energy was -0.41 eV/H2. It indicates thegravimetric density of 7.7 wt%, exceeding the U.S. Department of Energy's target of 6.5 wt%. According to estimates, a hydrogen fuel cell vehicle with a 6.5 wt% hydrogen storage density would have a range of 500 miles. Increasing the hydrogen storage density to 7.7 wt% would result in a range of approximately 590 miles (950 km).
Using the Van't Hoff equation, it was calculated that the average hydrogen decomposition temperature of TIG is 524 K (253 degrees Celsius). The electronic structure analysis results indicate that the hydrogen molecules are Kubas-type bonded to TIG. Upon adsorption, the hydrogen molecules acquire a net charge, which leads to a lengthening of the H-H bond distance but does not separate the molecule from its structure. When hydrogen is stored in TIG, the density of electronic states near the Fermi energy level in the 3d orbitals of titanium atoms is reduced compared to pure TIG. This suggests that after adsorption, the charge is transferred from the 3d orbitals of the titanium atoms to the 1s orbitals of the hydrogen molecules. When additional hydrogen molecules are incorporated into the system, the electrons in the 3d orbitals of the titanium atoms are further transferred to the 1s orbitals of the hydrogen, forming a stable adsorption.
To evaluate the structural thermal stability of the TIG materials, the team investigated the migration ability of the modified titanium atoms using the CI-NEB method. The diffusion migration barrier of titanium atoms was found to be 5.0 eV. At the average desorption temperature of 524 K, the thermal energy of titanium atoms was 0.68 eV, which was significantly lower than the diffusion energy barrier value. The migration of titanium atoms is limited because of the stable structural configuration and high energy barrier value at the reaction temperature. This prevents titanium atoms from migrating during hydrogen uptake and discharge, which avoids the destruction of the hydrogen storage structure caused by metal agglomeration. The team determined the thermodynamic stability of the material at both room temperature (300 K) and high temperature (600 K) using first-principles molecular dynamics simulations. The relevant results suggest that the TIG system is a viable hydrogen storage medium. These theoretical predictions offer a new option for developing efficient hydrogen storage materials.
The research results are published in the International Journal of Hydrogen Energy with the title "Stable and 7.7 wt% hydrogen storage capacity of Ti decorated Irida-Graphene from first-principles calculations". Yongkang Tan, a doctoral student at the School of Physics Science and Technology, Guangxi University, is the first author of the paper. Prof. Qing Peng from the Institute of Mechanics, Chinese Academy of Sciences and Prof. Yifang Ouyang from Guangxi University are the corresponding authors. This work was supported by the Liying Programm and the National Natural Science Foundation of China.
Details of the paper: Yongkang Tan, Xiaoma Tao, Yifang Ouyang, Qing Peng, “Stable and 7.7 wt% hydrogen storage capacity of Ti decorated Irida-Graphene from first-principles calculations”, International Journal of Hydrogen Energy, Volume 50, Part D, 2 January 2024, Pages 738-748.
Link to the paper: https://doi.org/10.1016/j.ijhydene.2023.08.115
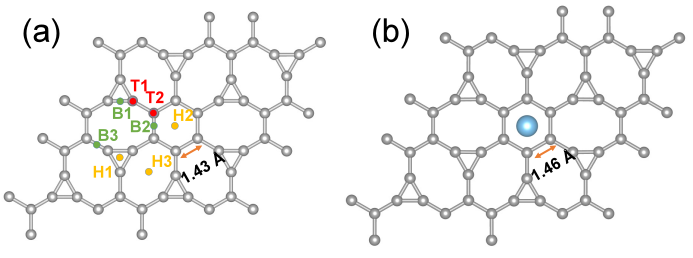
Fig. 1 Atomic structures of (a) 2![]() 2 supercell of Irida-Graphene and (b) Ti-decorated Irida-Graphene where Ti is positioned on the top of the hexagon ring (H2). The gray and blue spheres denote C-atoms and Ti-atoms, respectively. The hollow, top and bridge sites, the possible site for binding of Ti atom and Irida-Graphene, are marked in yellow, red, and green, respectively. After the Ti atom is decorated above the hexagonal carbon ring, the C-C bond of the hexagonal ring increases from 1.43 ? to 1.46 ?.
2 supercell of Irida-Graphene and (b) Ti-decorated Irida-Graphene where Ti is positioned on the top of the hexagon ring (H2). The gray and blue spheres denote C-atoms and Ti-atoms, respectively. The hollow, top and bridge sites, the possible site for binding of Ti atom and Irida-Graphene, are marked in yellow, red, and green, respectively. After the Ti atom is decorated above the hexagonal carbon ring, the C-C bond of the hexagonal ring increases from 1.43 ? to 1.46 ?.
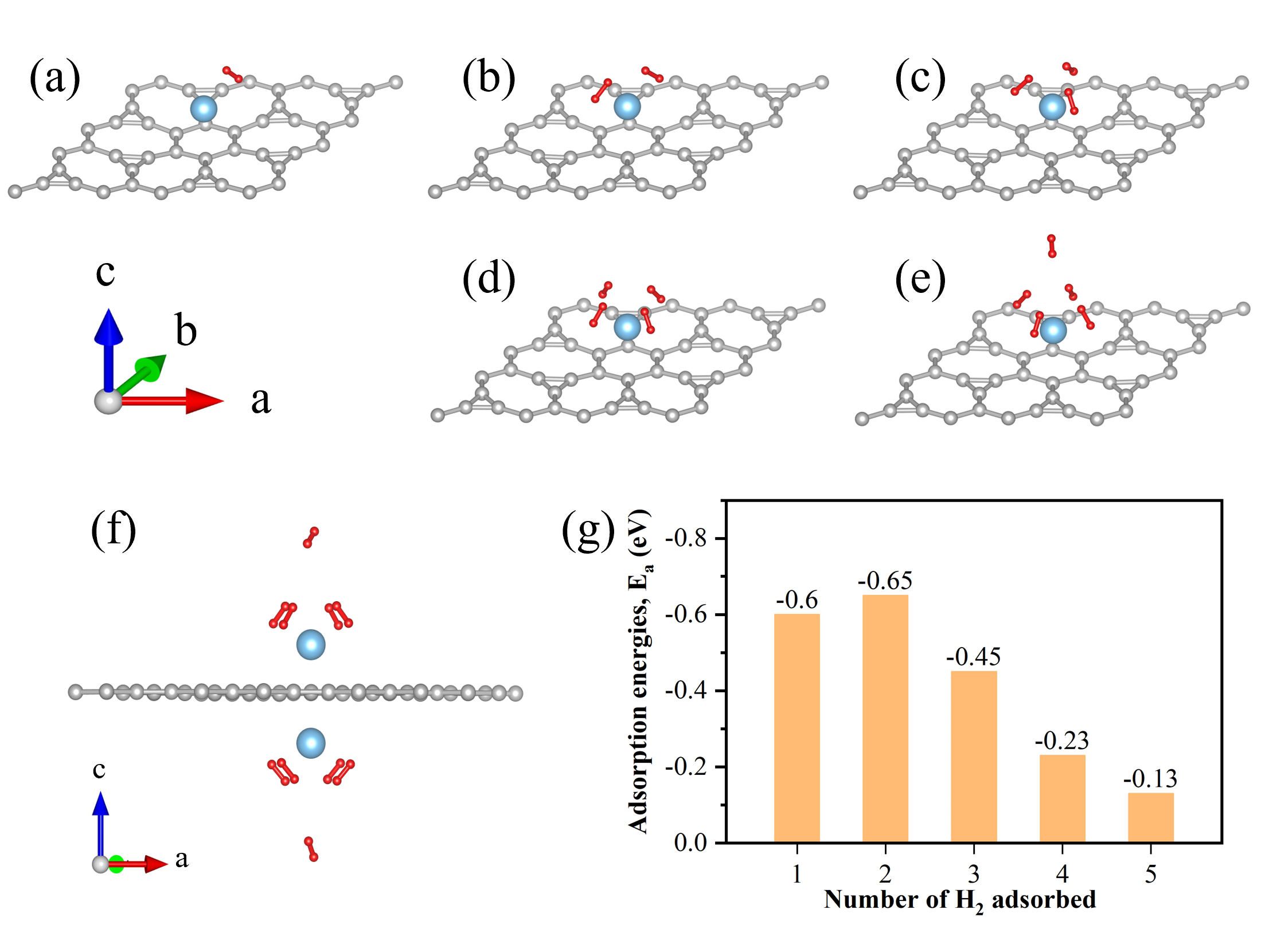
Fig. 2 Optimized structure and adsorption energies of H2 molecule being adsorbed on Ti-doped Irida-Graphene. (a)~(e) The structure of molecular adsorption of 1~5 H2. (f) The structure with a Ti atom placed on both sides of the Irida-Graphene, and each Ti atom absorbs five H2. The gray, blue and red balls represented C, Ti and H atoms, respectively. (g) The adsorption energies values for different H2 adsorption.
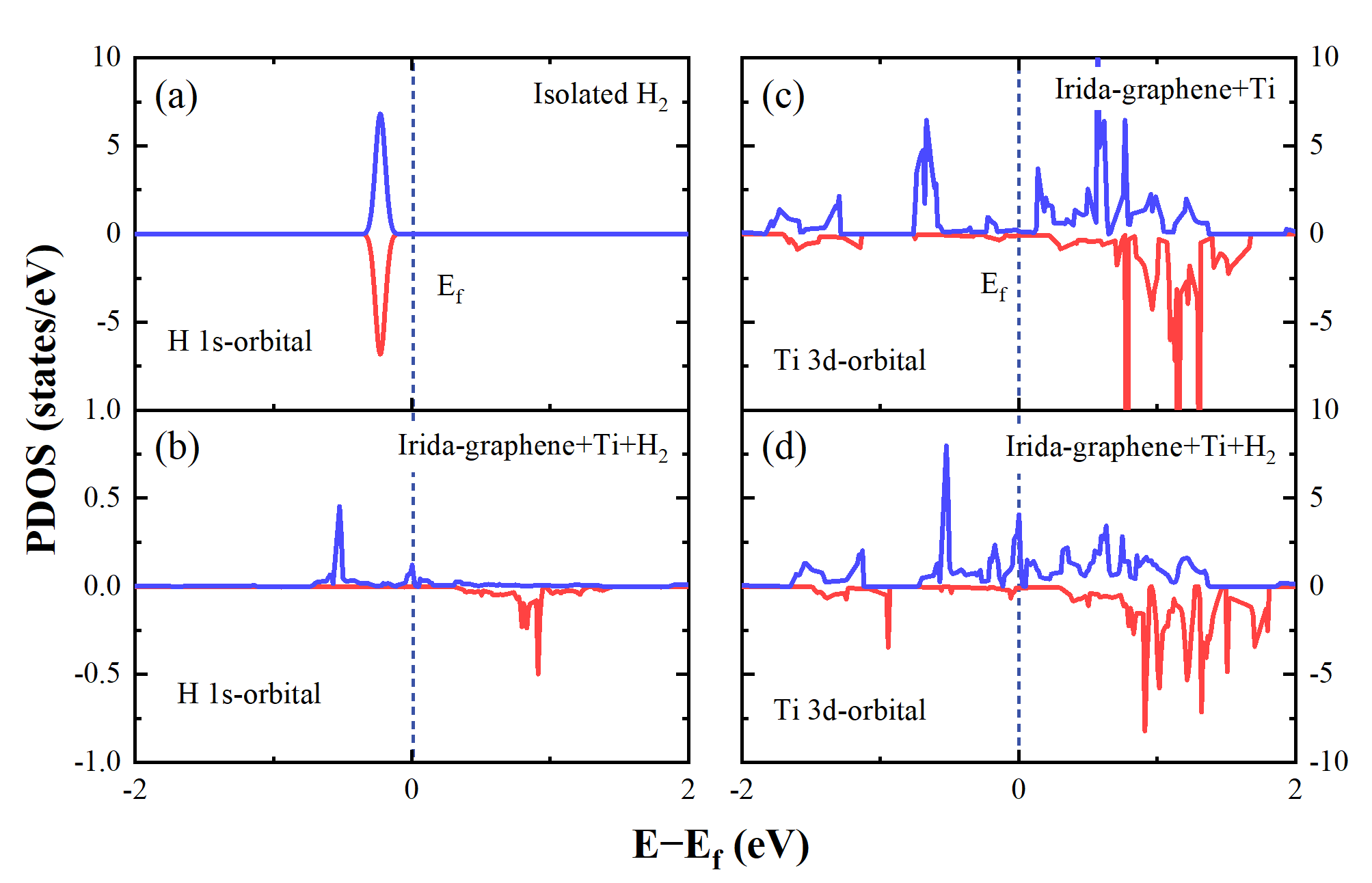
Fig. 3 Projected Density of States (PDOS) of H 1s-orbital and Ti 3d-orbital. (a) H 1s-orbital of isolated H2, (b) H 1s-orbital of Irida-Graphene + Ti + H2 system, (c) Ti 3d-orbital of Irida-Graphene + Ti and (d) Ti 3d-orbital of Irida-Graphene + Ti + H2. After the system absorbs H2, H 1s-orbital increased density near the Fermi level. Ti 3d-orbital shows a decrease below the Fermi level.
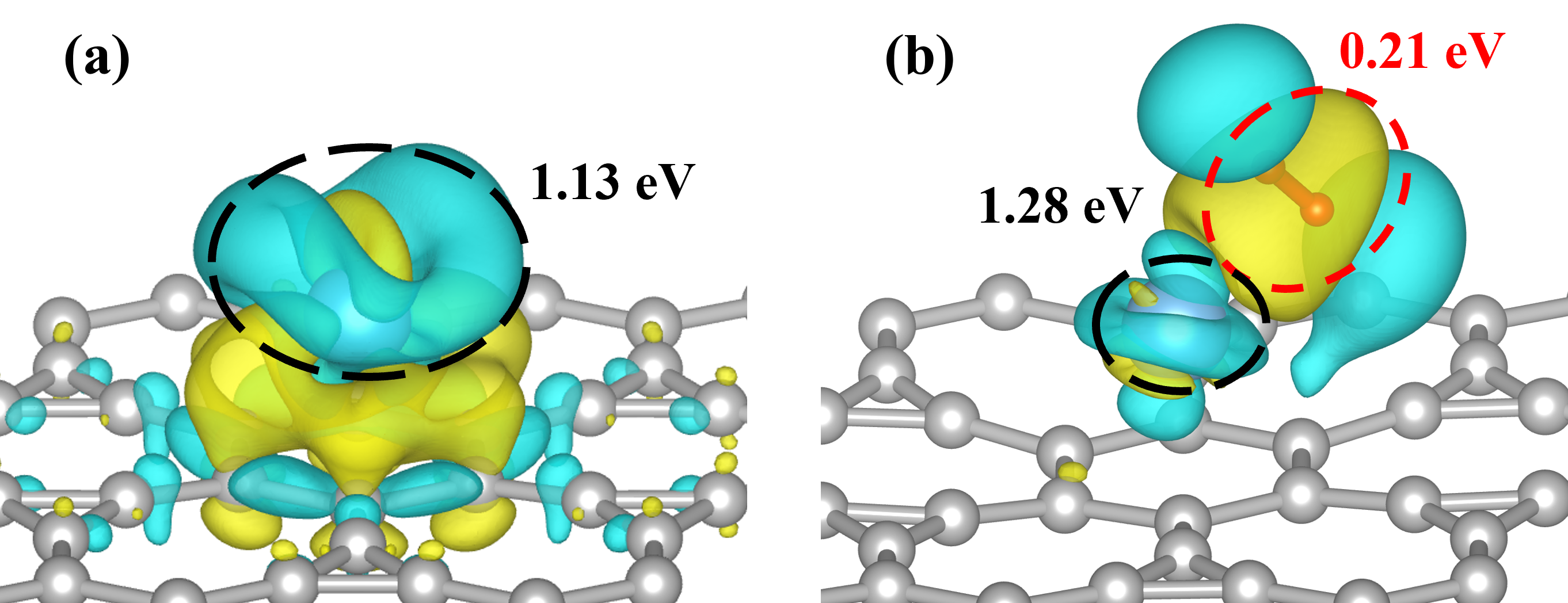
Fig 4 The different charge density of the system of Irida-Graphene + Ti (a) before and (b) after adsorption of first hydrogen molecule. The yellow color in the charge density map represents a higher concentration of electrostatic potential, while the blue color signifies the scarce concentration. The Bader charge within the atoms in dashed box was measured and recorded.
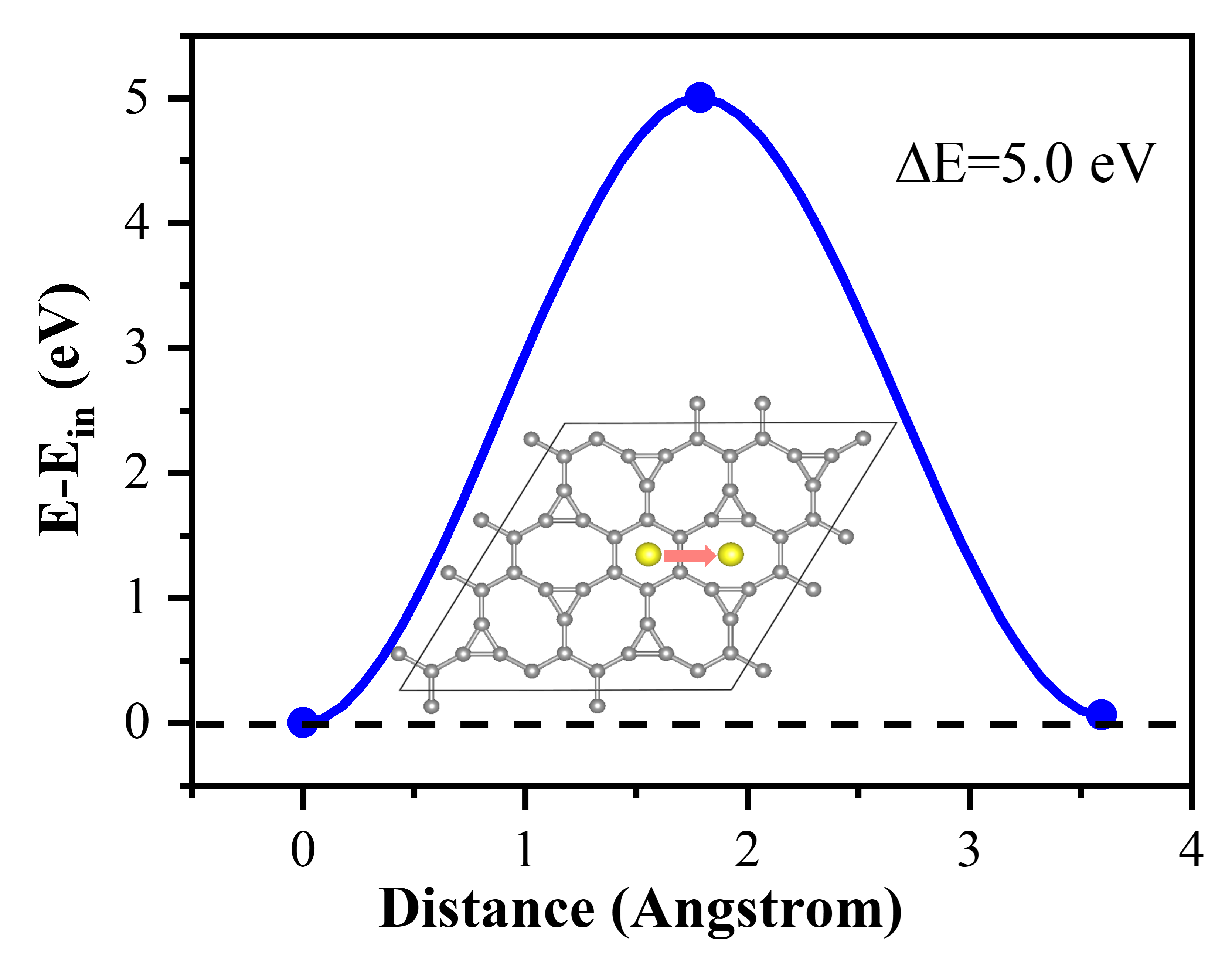
Fig. 5 Diffusion energy barrier for Ti atom migrates from hexagonal to the octagonal ring. The inset displays the diffusion path. The energy of the initial state was set to 0 eV and the final state is 0.07 eV higher than the initial state. The diffusion energy barrier is 5.0 eV along this diffusion path.
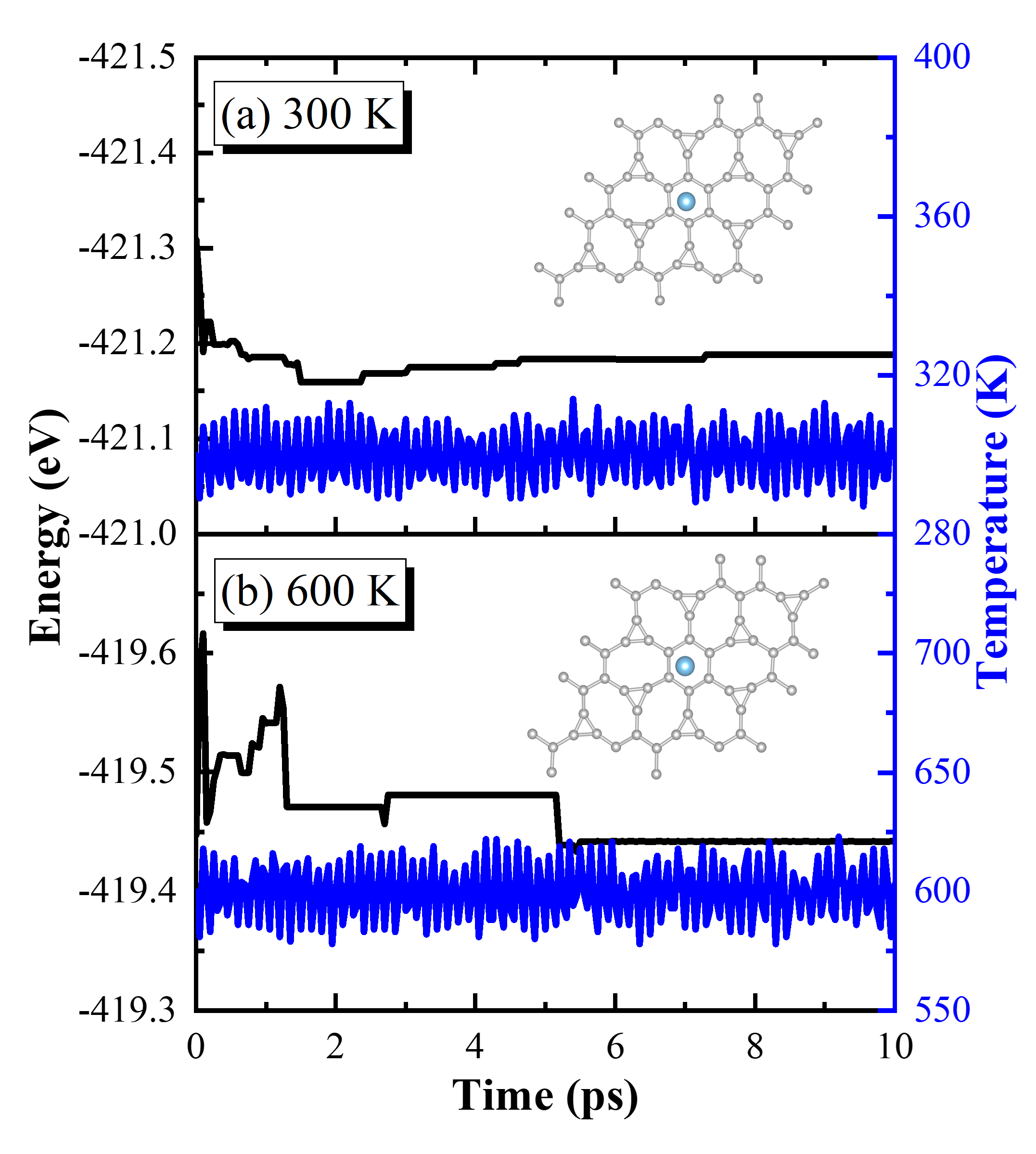
Fig. 6 Total energy and temperature of Irida-Graphene + Ti as a function of simulation time at temperature of (a) 300 K and (b) 600 K in AIMD with the canonical (NVT) ensemble. The insets in (a) and (b) are the AIMD snapshots of atomistic geometry of Irida-Graphene + Ti after 10 ps at 300 K and 600 K, respectively.